
Avec ses 1872 lits et places d’hospitalisation, le Centre Hospitalier Universitaire de Nice est un établissement de santé d’excellence au service de la population. Il offre une filière de prise en charge complète et diversifiée et peut répondre aux situations d’urgence.
L'UET du service de Dermatologie-Vénérologie possède une grande expérience des essais cliniques de phase II, III et IV, nationaux et internationaux dont il est souvent le centre coordonateur. Les domaines de compétence sont les suivants : psoriasis, eczéma atopique, cancers cutanés (mélanomes et carcinomes) et lésions pré-cancéreuses, acné, rosacée, vitiligo, pelade, maladie de Verneuil, dermatite séborrhéique, herpès, zona, dermatoses infectieuses (cellulite bactérienne…), mycoses cutanéo-muqueuses, prurit, photovieillissement, lésions pigmentées.
Un essai clinique est une recherche réalisée sur la personne humaine afin d’améliorer les connaissances biologiques ou médicales et ainsi optimiser la prise en charge des patients.
La recherche clinique est une activité hautement règlementée : la loi française, les règlements européens encadrent tous les aspects et étapes des essais cliniques. C’est une étape clé du développement du médicament. En partenariat avec les hôpitaux et l’industrie pharmaceutique, elle permet un accès aux thérapies les plus innovantes et contribue au niveau élevé d’expertise médicale des centres hospitaliers français.
Afin de garantir la sécurité des personnes se prêtant à la recherche et la validité des résultats, les essais cliniques sur le médicament se déroulent en plusieurs phases.
La participation à un essai clinique doit être libre et éclairée. Chaque étape de l’essai doit vous être expliquée de manière claire et détaillée. Vous devez être conscient(e) des avantages et des potentiels inconvénients à participer à un essai clinique. La participation à l’essai ne pourra se faire qu’une fois votre consentement recueilli.
Le bénéfice thérapeutique du patient est l’objectif premier du déploiement des essais cliniques dans les centres hospitaliers.
Toute personne peut participer à un essai clinique, à condition de correspondre aux critères de sélection d’entrée dans l’étude. Ces critères varient selon les études et sont définis pour assurer la sécurité des patients et garantir la cohérence et la solidité des résultats de l’étude.
Seul le médecin responsable de l’essai clinique sera en mesure de vous dire si vous pouvez participer ou non à l’étude.
L’ANSM (Agence Nationale de Sécurité du Médicament) vérifie la sécurité du produit expérimental ainsi que des actes prévus dans le protocole afin de ne pas exposer les participants à un risque inconsidéré. L’ANSM est une organisation publique dépendante du ministère de la Santé. Elle est responsable de la mise sur le marché et de la surveillance d’utilisation des produits de santé. Dans le cadre des essais cliniques et elle délivre les autorisations de recherche clinique menées en France. https://ansm.sante.fr
Le CPP (Comité de Protection des Personnes) est chargé de vérifier que le protocole de recherche est conforme aux règles éthiques, médicales et juridiques afin d’assurer la protection du participant à l’essai. Les membres du CPP sont bénévoles, tenus au secret professionnel.
Le médecin investigateur
Dans le cadre des essais cliniques, seul un médecin qualifié est autorisé à être investigateur. C’est le seul à pouvoir proposer à un patient de participer à un essai clinique. Il doit posséder un niveau de formation et d’expertise suffisant lui donnant droit d’exercer au sein d’une unité de recherche clinique. Il doit également avoir une connaissance de la réglementation des essais cliniques. Il assure le suivi médical des patients inclus dans l’essai clinique selon le protocole de surveillance prédéfini par l’étude et est chargé de leur sécurité. Il pourra tout au long de l’étude répondre à vos questions concernant l’essai clinique.
L’Attaché de Recherche Clinique (ARC)
L’ARC aide les investigateurs à gérer le côté pratique des essais cliniques dans le centre. Il connait parfaitement la réglementation des essais cliniques. L’ARC veille au respect des droits des patients, du protocole de l’étude, des Bonnes Pratiques Cliniques et des procédures opératoires standard de l’essai. Il assure également la communication entre le laboratoire portant l’essai et l’équipe médicale investigatrice.
L’infirmier de Recherche clinique
L’IRC est très impliqué dans les essais cliniques. Il aide à la réalisation et/ou à la préparation aux examens nécessaires pour l’étude. Il est en permanence au contact du patient et transmet régulièrement et rapidement aux médecins investigateurs et aux attachées de recherche clinique toute information importante concernant le patient.
Le Pharmacien clinique
Le pharmacien veille à la bonne dispensation du traitement. Il a pour mission principale d’assurer la gestion pharmaceutique des produits de santé expérimentaux (médicaments et dispositifs médicaux) pour l’ensemble des essais cliniques menés par les médecins investigateurs.
Organisme public dépendant du ministère de la Santé responsable de la mise sur le marché et de la surveillance d’utilisation des produits de santé.
Dans un essai clinique, il doit veiller au respect des droits des patients, du protocole de l’étude, des Bonnes Pratiques Cliniques et des procédures opératoires standard de l’essai.
Autorisation administrative de commercialisation d’un médicament délivrée sur avis de l’Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM) à l'établissement pharmaceutique le produisant.
Ensemble de dispositions ayant pour but de concourir à la protection des droits, à la sécurité et à la protection des personnes se prêtant à ces recherches ainsi qu’à la crédibilité et à la confidentialité des données à caractère personnel et des résultats de ces recherches.
Amélioration de l’état de santé ou du bien-être d’une population de patients soumis à un traitement ou une stratégie thérapeutique.
Base de données en ligne qui fournit des informations sur des études cliniques sur une large gamme de maladies.
Les membres de CPP sont bénévoles, tenus au secret professionnel, indépendants vis à vis des investigateurs et des promoteurs et désignés par l’autorité administrative compétente. Le CPP veille à la protection des personnes qui se prêtent à une recherche médicale et veille au respect de la législation dans le cadre de la recherche médicale. L’avis favorable d’un CPP est obligatoire avant le début d’une recherche portant sur la personne humaine. Cet avis est demandé par le promoteur de la recherche auprès d’un CPP. Un seul avis est demandé pour chaque recherche. En cas de modification substantielle en cours de la recherche, une demande d’avis sur cette modification doit être envoyée au CPP.
Commission veillant à la protection de la vie privée et des libertés des personnes notamment au regard du traitement des données dans le cadre des recherches.
Acceptation libre et formellement exprimée d’une personne en vue de participer à une recherche dont elle a été informée des objectifs et contraintes.
Critère d’évaluation permettant de répondre à l’objet principal de l’étude.
Critères définissant les personnes non éligibles pour une recherche clinique.
Critères définissant les caractéristiques des sujets ou des patients qui peuvent participer à une étude.
Critères permettant de documenter les objectifs secondaires fixés dans l’étude.
Délivrance d’un médicament à titre clinique ou dans une recherche clinique.
Procédure expérimentale où ni le participant ni l'équipe de recherche ne connaissent le groupe de traitement auquel appartient chaque participant.
Temps séparant la délivrance de l’information et la signature du consentement ou l’expression de la non-opposition.
Toute réaction nocive, survenant chez une personne traitée par un médicament et attribuable à ce dernier.
Effet indésirable qui nécessite l’hospitalisation, met la vie en danger, provoque un handicap ou une malformation congénitale. Il doit impérativement être immédiatement reporté à l’ANSM.
Essai clinique dans lequel l'investigateur et/ou la personne qui y participe connaissent le traitement qui est donné à cette personne lors de l’étude. Inverse : essai en insu.
Essai dont les traitements sont attribués par tirage au sort (randomisation).
Processus établissant la participation d’une personne à une recherche clinique. L’inclusion d’un participant repose à la fois sur son éligibilité et son consentement éclairé.
Façon de garantir que les personnes impliquées dans une étude de recherche, comme les participants à des essais cliniques, ne savent pas à quel groupe de l'essai ils appartiennent.
Médecin qui dirige et surveille la réalisation de la recherche portant sur la personne humaine (article L1121-3 du Code de la Santé publique).
Investigateur responsable d'une équipe d'investigateurs chargée de la conduite d'un essai clinique sur un site d'essai clinique.
Lieu disposant des moyens humains, matériels et techniques adaptés à la recherche et compatibles avec les impératifs de sécurité des personnes qui s'y prêtent.
Changement apporté à n'importe quel aspect de l'essai clinique susceptible d'avoir une incidence substantielle sur la sécurité ou les droits des participants ou sur la fiabilité et la robustesse des données obtenues lors de l'essai clinique.
Substance ou composition présentée comme ayant des propriétés curatives ou préventives à l'égard des maladies humaines, ainsi que tout produit pouvant être administré à l'homme en vue d'établir un diagnostic médical ou de restaurer, corriger ou modifier ses fonctions organiques.
Un médicament biologique, ou biomédicament est un médicament produit par un organisme vivant, spontanément ou par génie génétique, par opposition aux substances produites par voie chimique, souvent qualifiées de petites molécules. Grâce aux progrès du génie génétique, les biomédicaments occupent une part croissante de l'arsenal thérapeutique : hormones comme l’insuline, antigènes monoclonaux, enzymes de substitution, etc.
Médicament expérimenté dans une recherche portant sur la personne humaine.
Document écrit qui doit être remis par l'investigateur à la personne dont le consentement à participer à une recherche est sollicitée résumant les informations sur celle-ci. (article L122-1 du Code de Santé Publique) Ce document doit être approuvé par le CPP. L'information porte notamment sur le but et le déroulement de l’essai, les éventuels bénéfices et/ou risques attendus, la forme de l’étude et les traitements étudiés, etc.
Respect par un sujet ou un malade des règles de prescription d’un médicament.
Une personne, volontaire sain ou malade, prenant part à un essai clinique, qu'elle reçoive un médicament expérimental ou qu'elle serve de témoin.
Participant à un essai clinique et dont l’investigateur n’a plus de nouvelles. Plus le nombre de participants perdus de vue dans un essai est important, plus l’imprécision est grande sur les résultats de l’essai. Si cette sortie est liée à un effet lié aux traitements, les perdus de vue peuvent introduire un biais dans l'étude.
Branche de la pharmacologie qui étudie le sort du médicament dans un organisme vivant, c’est-à-dire l'effet de l'organisme sur le médicament. La pharmacocinétique étudie en termes de quantité et de rapidité le comportement de l’organisme vis-à-vis du médicament : son absorption, son éventuelle fixation sur des protéines du sérum, sa distribution dans les différents tissus, ses possibles modifications (métabolisme) et son excrétion par l’organisme.
Branche de la pharmacologie qui étudie l’ensemble des effets induits par un médicament sur un organisme vivant.
Branche de la pharmacologie ayant pour objet la surveillance, l’évaluation et la prévention du risque d’effets indésirables résultant de l’utilisation des médicaments. La pharmacovigilance se différencie de la toxicologie en ce qu'elle se réalise en situation réelle, c’est-à-dire après l’obtention de leur AMM.
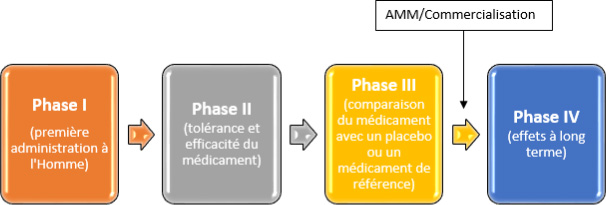
Phases systématisées dans la mise au point clinique d’un médicament.
Le développement d’un nouveau médicament chez l’homme est décrit en 3 phases
successives à l’issue desquelles pourra être demandée l’autorisation de mise sur le marché
(voir AMM) et une phase lui succédant.
- Phase I : étudie la tolérance, la pharmacocinétique et la pharmacodynamie du nouveau
médicament chez l’homme. Les essais de phase I se déroulent le plus souvent chez des
volontaires sains sauf quand le traitement étudié présente une toxicité trop importante pour
être administré chez des volontaires sains (principe éthique de bienfaisance). C’est le cas
notamment en cancérologie .
- Phase II : étudie la relation existant entre les doses du médicament et les effets
thérapeutiques (bénéfiques) ou indésirables (toxiques) du nouveau médicament chez des
malades dans l’indication envisagée du médicament.
- Phase III : étudie le rapport entre l’efficacité et la tolérance du nouveau médicament chez un
grand nombre de patients exposés au médicament pendant des durées variables selon la
pathologie et le mode de prescription futur du médicament, dans les conditions envisagées
d’utilisation.
- Phase IV : est une phase de pharmacovigilance, qui se situe après introduction sur le marché
et étudie la survenue d’éventuels effets indésirables, notamment rares, en condition normales
d’utilisation.
La systématisation de la mise au point d’un médicament en phases successives peut être
assouplie dans certaines situations. Ainsi la phase I de médicaments dotés d’une toxicité
propre ou ne manifestant leur effet que chez des malades sera réalisée sur des malades. Dans
les pathologies concernant un petit nombre de sujets, et donc de potentiels participants, les
phases II et III peuvent être réunies (phase II-III).
Substance sans effet pharmacologique démontré utilisée dans le cadre des essais cliniques comme comparateur du médicament étudié. Le but est de distinguer l’effet pharmacologique propre d’une substance de l’effet suggestif produit par l’administration d’un produit quel qu’il soit (effet placebo), lié à la confiance qu’a le patient dans des effets de ce produit. Le Placebo a la même forme, la même couleur, la même odeur que celles du médicament étudié mais ne contient pas de substance active.
Ensemble des individus participant à une étude. Dans un essai clinique, les critères d'inclusion et d'exclusion décrivent à qui pourra ou ne pourra pas être proposé de participer, et par conséquent définissent les caractéristiques de la population de l'étude.
Principes philosophiques et moraux visant à protéger les participants à la recherche clinique et à garantir l'intégrité de la recherche. Initialement portés par des textes déclaratifs (Code de Nüremberg, Rapport Belmont, Déclaration d’Helsinki, Convention d’Oviedo) ils sont aujourd’hui à la base de réglementations, soit en étant explicitement contraignantes, soit par référence aux textes déclaratifs. Ces différents textes mettent en avant trois principes éthiques majeurs : la Bienfaisance ou Non Malfaisance, l’Autonomie ou respect de la personne et la Justice distributive.
Personne physique ou morale qui prend l’initiative d’une recherche portant sur la personne humaine, en assure la gestion et vérifie que son financement est prévu. En pratique, le promoteur doit choisir l’investigateur, recruter des ARC, soumettre le protocole de la recherche à l’avis du CPP et à l’ANSM, contracter une assurance couvrant les conséquences éventuelles de cette recherche, et déclarer à l’ANSM, au CPP et aux investigateurs les événements indésirables graves inattendus survenus au cours de la recherche. De plus, il est habituel que le promoteur prenne en charge les coûts de la recherche mais ce n’est pas obligatoire puisqu’un tiers peut financer la recherche sans se porter obligatoirement promoteur. A la fin de la recherche, le promoteur avise l’autorité compétente et le CPP que la recherche est terminée, indique les raisons qui motivent l’arrêt de la recherche lorsqu’il est anticipé Terme anglais : sponsor.
Document rassemblant tous les éléments descriptifs d’une recherche portant sur la personne humaine et qui précise les conditions dans lesquelles cette recherche doit être réalisée et gérée. Ce document daté, approuvé par le promoteur et par l’investigateur, est soumis au CPP et à l’ANSM, en intégrant le cas échéant, les modifications successives. Le terme « protocole» recouvre les versions successives du protocole ainsi que ses modifications.
Mesure du bien être initialement utilisée en économie de la santé. Elle exprime l'effet de facteurs tels que les symptômes, la douleur, la santé psychologique et le bien-être sur la vie des gens. Les mesures de qualité de vie liée à la santé peuvent être utilisées pour calculer l'impact des traitements sur les vies des patients et à ce titre faire partie des critères d’évaluation d’une recherche clinique.
Rapport théorique qui existe entre le bénéfice thérapeutique attendu du traitement et le risque potentiel d’effets indésirables de ce traitement.
Recherches organisées et pratiquées sur l'être
humain en vue du développement des connaissances biologiques ou médicales.
Il existe trois catégories de recherches impliquant la personne humaine :
1° Les recherches interventionnelles qui comportent une intervention sur la personne non
justifiée par sa prise en charge habituelle ;
2° Les recherches interventionnelles qui ne comportent que des risques et des contraintes
minimes, dont la liste est fixée par arrêté du ministre chargé de la santé, après avis du
directeur général de l'Agence nationale de sécurité du médicament et des produits de santé ;
3° Les recherches non interventionnelles qui ne comportent aucun risque ni contrainte dans
lesquelles tous les actes sont pratiqués et les produits utilisés de manière habituelle.
(Article1121-1 du Code de Santé Publique).
Individu autorisé (par exemple par un tribunal) à prendre des décisions au nom d'une autre personne. Dans des circonstances où un patient n'est pas en mesure de donner lui-même un consentement éclairé (par exemple, des patients en soins intensifs), un représentant légal peut parfois donner un consentement ou une autorisation en son nom, y compris concernant des traitements médicaux.
Probabilité qu’un préjudice se produise lors de l’utilisation d’un traitement dans la pratique clinique (risque thérapeutique) ou dans le cadre d’une recherche (risque expérimental). Le préjudice peut être physique, psychologique, social ou économique. Aucun essai clinique ne peut être réputé sans risque. Avant de décider de prendre part ou non, les participants doivent être conscients à la fois des bénéfices et des risques (voir consentement éclairé). Dans un essai clinique les risques peuvent avoir différentes origines : effets secondaires d’un traitement, attribution d’un médicament moins efficace que le traitement de référence par exemple. Les risques liés à des effets secondaires imprévus sont plus importants dans les premiers stades des essais cliniques.
Mode de décision reposant sur le hasard. L’attribution d’un traitement à une personne se prêtant à la recherche par tirage au sort. Elle permet de réduire les biais résultant de l’attribution d’un traitement donné aux participants présentant certaines particularités (âge, stade de la maladie, genre). Le tirage au sort concerne le traitement reçu ou l’ordre des administrations dans les études en cross-over. La procédure de tirage au sort est généralement couplée à celle d’insu, l’administration « en ouvert » d’un comparateur, notamment un placebo pouvant être psychologiquement difficile Dans le cadre de la recherche clinique, le terme anglais de randomisation est le plus couramment employé.
Capacité de l'organisme à supporter une certaine substance. On désigne parfois par « tolérance au médicament », la diminution de la réaction à des doses constantes répétées d'un médicament ou la nécessité d'augmenter les doses pour maintenir un effet constant. Cette « tolérance au médicament » peut entraîner une dépendance physique (physiologique) ou émotionnelle, qui est un état d'adaptation associé à un syndrome de sevrage à l'arrêt d'une exposition répétée à un médicament.
Degré auquel une substance chimique ou biologique peut nuire à un organisme vivant. Les effets néfastes peuvent concerner des organes, des tissus ou des cellules spécifiques ou l'organisme entier. Le développement des médicaments est un processus pas à pas qui implique l'évaluation de leur innocuité aussi bien pour les animaux que pour les êtres humains. Les études d'innocuité non cliniques (avant les essais sur l'homme) doivent permettre d'identifier les effets toxiques potentiels susceptibles de se produire dans les conditions de l'essai clinique ultérieur.
Traitement, reconnu par la communauté scientifique internationale et les autorités de santé comme le mieux établi scientifiquement dans une indication donnée. Il sert de comparateur avec la méthode de soin à l’étude ; celui-ci sera alors qualifié de plus, autant ou moins efficace et/ou de mieux, autant ou moins bien toléré.
Personne qui agit sans contrainte, selon son libre arbitre. Toute personne qui se prête à une recherche portant sur la personne humaine doit être volontaire au sens du Code de la Santé Publique ( art. L1122-1) "Préalablement à la réalisation d’une recherche portant sur la personne humaine, le consentement libre, éclairé et exprès de celle-ci doit être recueilli après que l’investigateur ait fourni les informations prévues par la loi".
Personne en bonne santé qui participe de son plein gré à une recherche clinique. Tout médicament est d'abord évalué chez le volontaire sain (cf. essai de phase I) à l'exception de médicaments aux effets indésirables lourds (Ex: oncologie) et lorsque l’éthique ne le permet pas. Dans ces cas le traitement est évalué chez des malades.

Investigateur principal : Pr. Thierry Passeron Co-Investigateurs : Dr. Dugourd et Dr. Philippo

Investigateur principal : Dr. Henri Montaudié
Co-Investigateurs : Dr. Picard-Gauci et Dr. Troin

